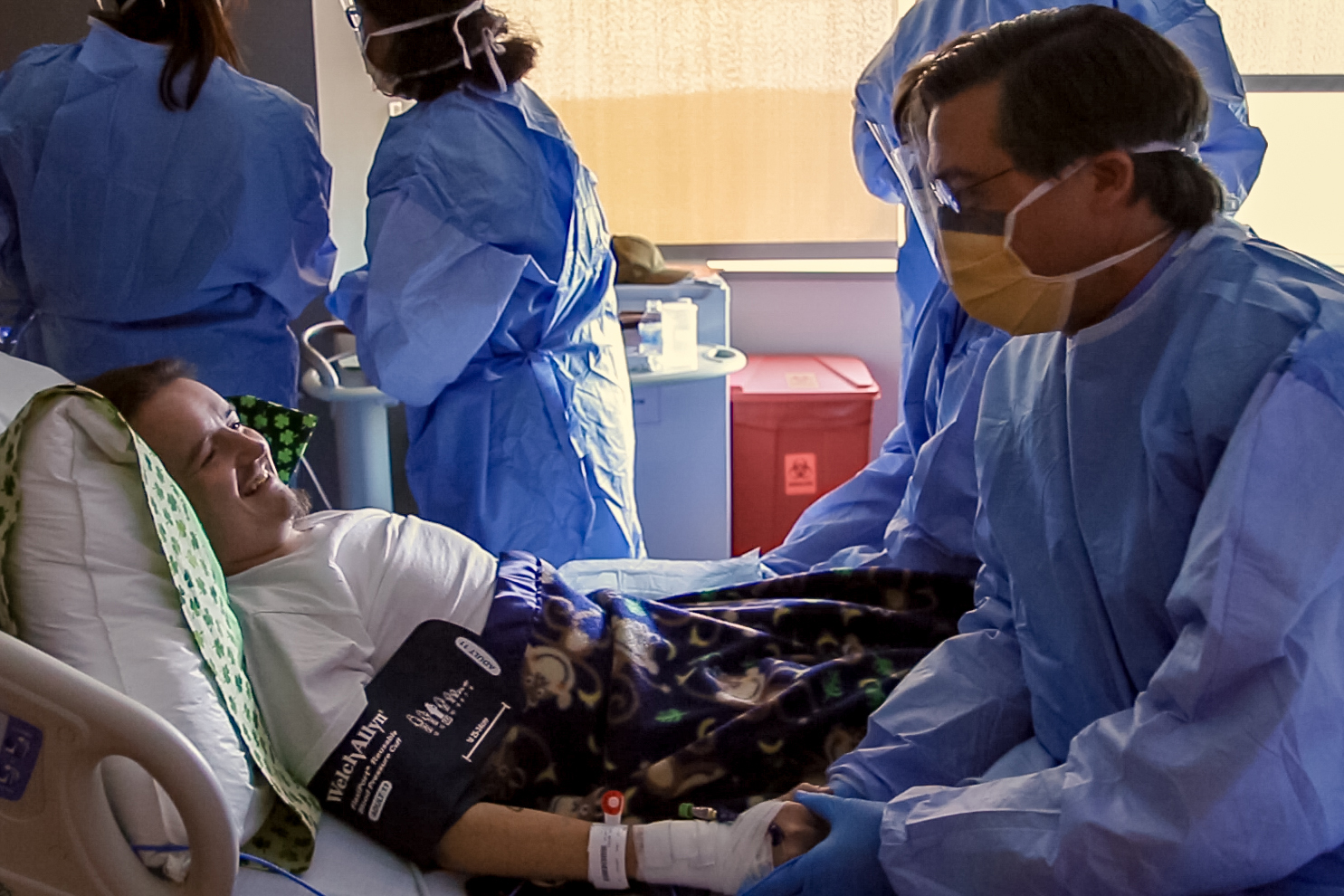
Dr. David Weinstein and his team at UConn Health have administered to a patient the world’s first investigational gene therapy for potentially deadly glycogen storage disease (GSD).
On July 24, the novel gene therapy to treat the rare childhood genetic disorder that impacts the liver’s storage and release of sugar was successfully given to the first human patient through a one-time, 30-minute infusion at UConn John Dempsey Hospital.
The gene therapy is carried through the patient’s bloodstream by a naturally occurring virus to the liver to replace deficient sugar enzymes caused by the disease and to jumpstart the body’s glucose control.
“Gene therapy is basically delivering a new copy of the gene to the liver,” Weinstein says.

GSD affects children from infancy through adulthood, causing dangerously low blood sugar levels and constant dependence on glucose consumption in the form of cornstarch every few hours in order to survive. Unfortunately, if a cornstarch dose is missed or a patient oversleeps, the disease can lead to seizures and death. Normally, healthy livers store excess sugar from food for the body’s future energy needs and release it into the bloodstream when needed in the form of processed sugar enzymes known as glycogen. In GSD, however, the liver fails to break down glycogen into glucose, causing the body’s blood sugar levels to drop.
“I am excited to finally see this dream become a reality with our first GSD patient receiving the promising gene therapy. We’ve been tirelessly working toward and anxiously awaiting this milestone for 20 years, and so have our patients,” says Weinstein, the world-renowned pediatric endocrinologist and scientist leading the clinical trial, who cares for 600 patients from 48 countries. Weinstein is director of the Glycogen Storage Disease Program at Connecticut Children’s Medical Center and UConn Health, the largest in the world for the care and treatment of this condition.
The ground-breaking clinical trial’s primary goal is testing the safety and dosage of the investigational gene therapy for those with GSD Type Ia. The first patient is being closely monitored following infusion, and within six weeks doctors plan to halt the patient’s daily regimen of cornstarch to test the effectiveness of the gene therapy. In animal models, the injection has already shown promise to be safe, effective, and long-lasting.
“Our GSD Program team and our patients around the U.S. and the world now have the hope they have been waiting for,” says Weinstein, “thanks to the start of this new gene therapy clinical trial. This is the first big leap forward for GSD.”
GSD has various forms (types 0, Ia, Ib, III, VI, IX, and XI) that affect one out of every 100,000 people. The clinical trial is initially focusing on GSD Type Ia, affecting an estimated 6,000 patients worldwide, which is caused by a defective gene for the enzyme glucose-6-phosphatase-α (G6Pase-α) that controls sugar release from the liver. The condition was almost always fatal until 1971, when it was discovered that continuous glucose therapy could help these patients. Cornstarch therapy was introduced as a slow release form of glucose in 1982, and it allowed feeds to be spaced to every three to four hours. Thanks to cornstarch, a greater number of patients with GSD are now surviving into adulthood.
The clinical trial is in conjunction with the biopharmaceutical company Ultragenyx based in Novato, California. The Phase 1/2 clinical trial, approved by the FDA in late April 2018, will soon expand from UConn Health in the U.S. to other sites including Canada, Spain, and the Netherlands.
The GSD Program’s multidisciplinary team at Connecticut Children’s provides comprehensive clinical care to patients, while the program’s research laboratories and clinical trial are located at UConn School of Medicine at UConn Health.
For more information visit the GSD Program’s website or call: 860-837-7800.