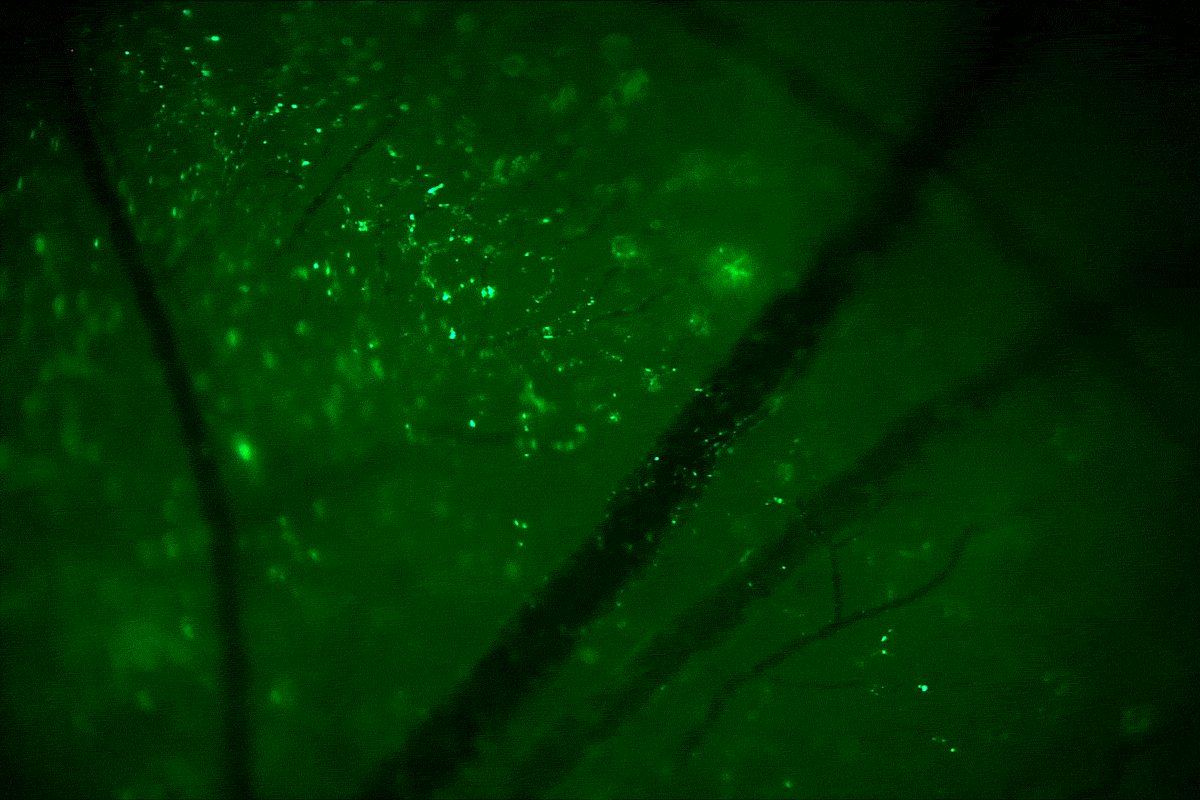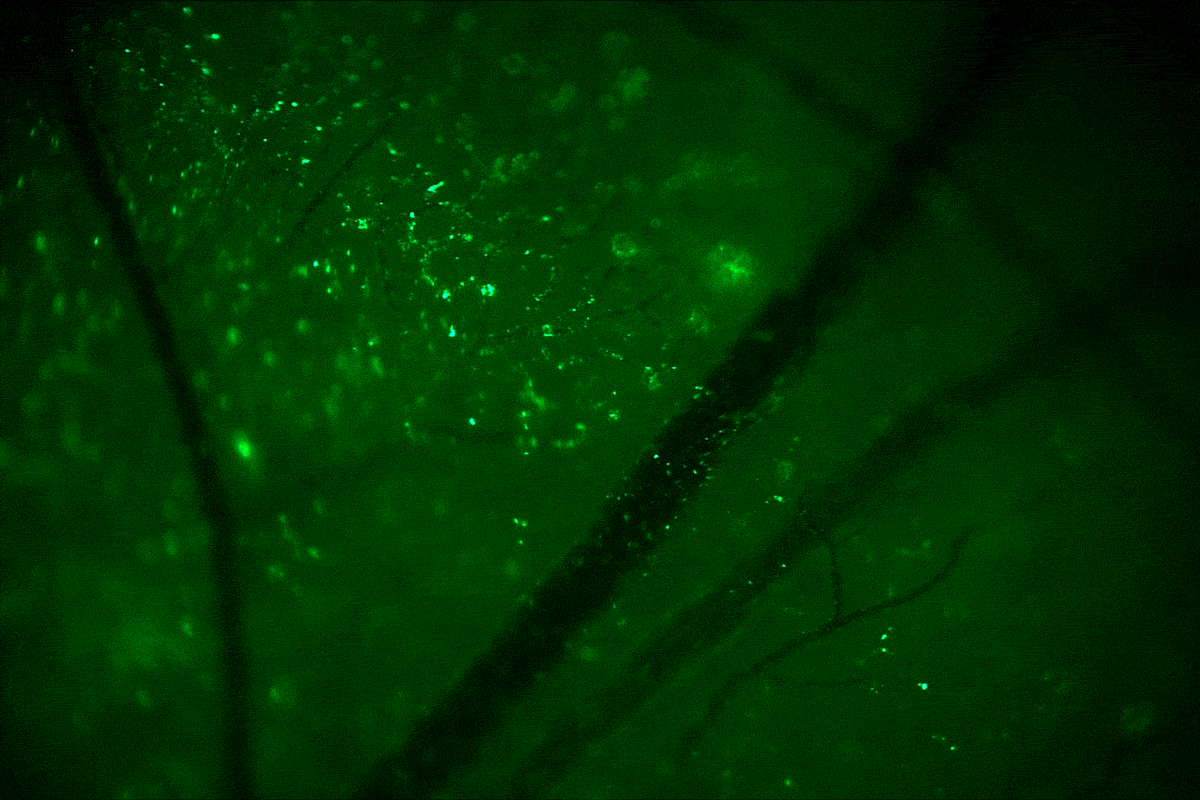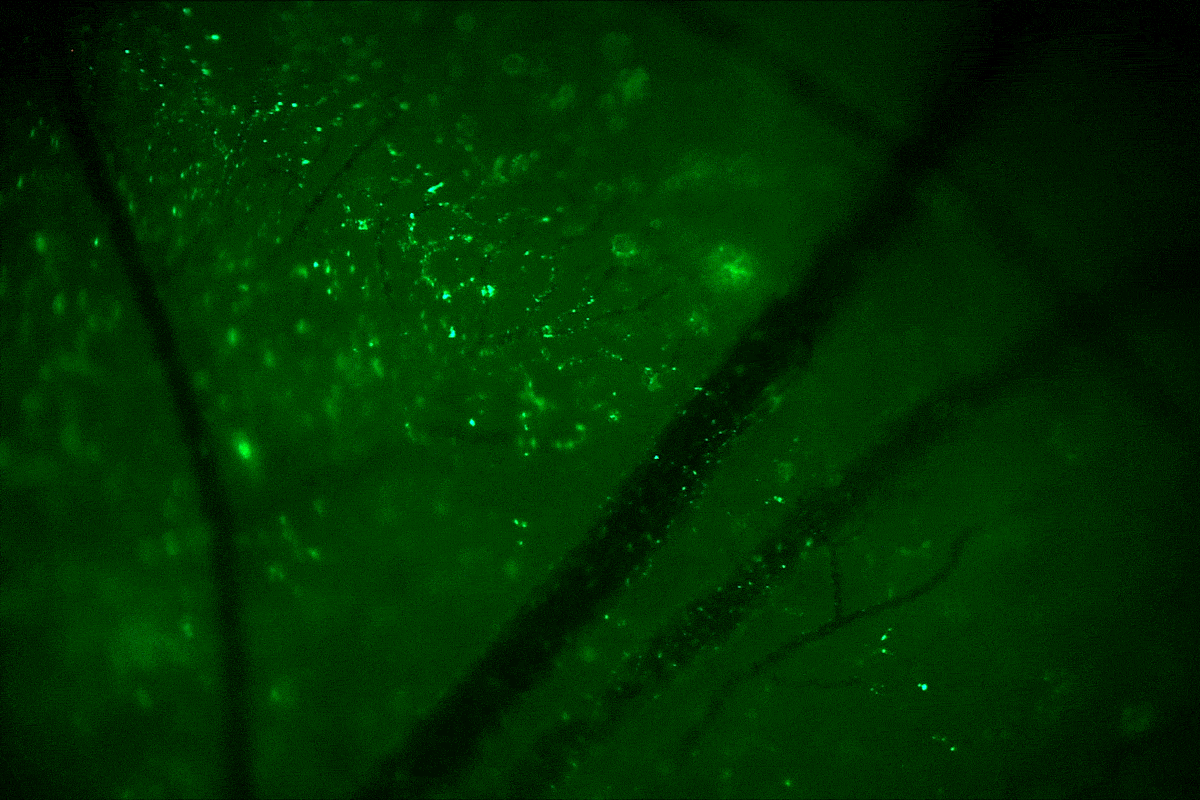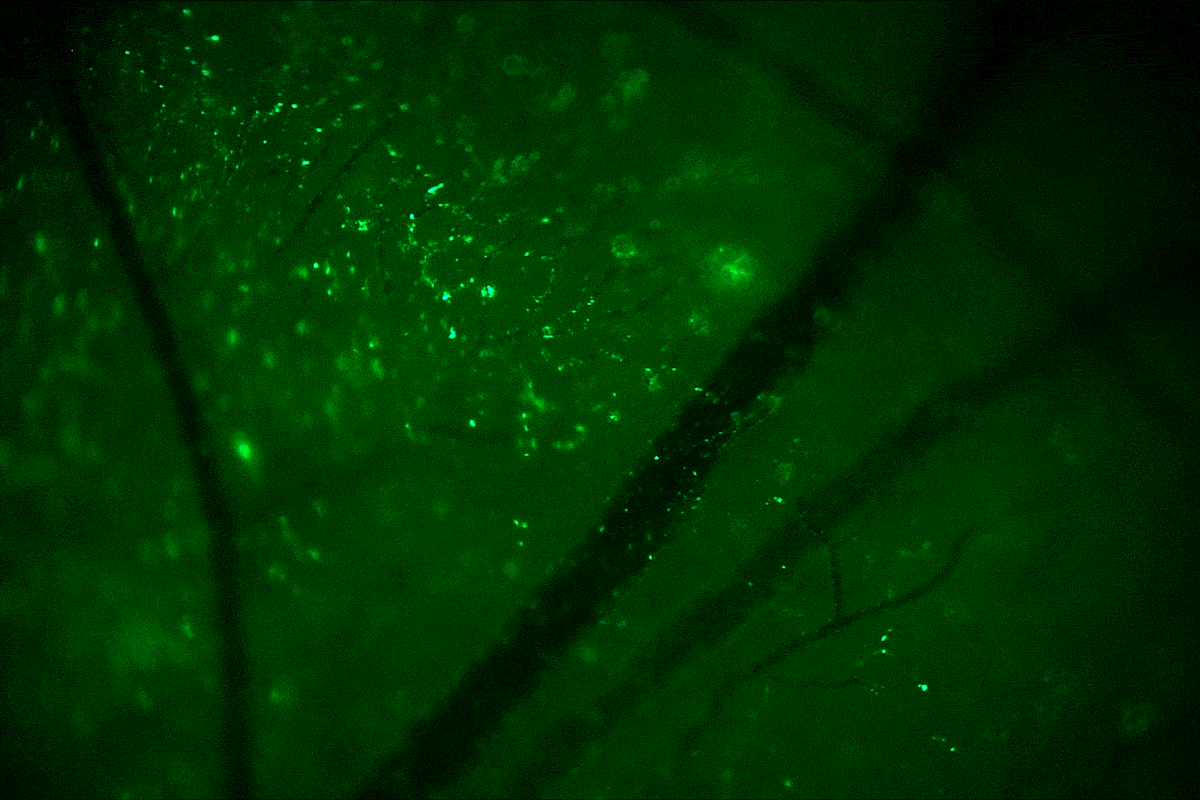
A team of researchers, including UConn assistant professor of pharmaceutics Raman Bahal, has, for the first time, corrected a genetic mutation in a mammalian fetus using a targeted gene editing technique. The approach offers a potential new pathway for treating inherited genetic disorders during the earliest stages of development.
Every year, an estimated 8 million children are born with severe genetic disorders or birth defects. While genetic conditions can be detected during pregnancy using amniocentesis, there are no treatment options currently to correct the conditions before birth.
Yet in a paper appearing in the international science journal Nature Communications, scientists from UConn, Yale University, and Carnegie Mellon University describe how they corrected a genetic mutation in fetal mice using an intravenous injection of nanoparticles loaded with a combination of donor DNA and synthetic molecules known as peptide nucleic acids or PNAs.
While the results are encouraging, the researchers caution that much more research needs to be done before the application can even be considered for human trials.
“This study provides a benchmark for gene editing technology where diseases can be treated at a very early stage of development,” says Bahal, a recent arrival to UConn who performed the research when he was an associate scientist working in the Yale lab of Dr. Peter Glazer, a professor of therapeutic radiology and genetics.
In the current study, the researchers injected the nanoparticle suspension into the amniotic fluid of pregnant mice whose fetuses carried a genetic mutation that causes beta thalassemia, a blood disorder that reduces the production of hemoglobin and causes a severe form of anemia. The nanoparticles deliver the PNA/DNA complex to the site of the mutation, where the PNA molecule, which mimics DNA, binds to the faulty gene’s DNA, creating a triple helix. This helical distortion activates the mutated cell’s repair response, which corrects the mutation using the healthy donor DNA packaged with the PNA.
Four months after birth, the treated mice showed dramatic improvements in symptoms of beta thalassemia, enough so for them to be considered cured. Mice that received a single injection of the PNA/DNA complex in utero had normal blood counts, spleens of normal size, and lived a normal life span. Untreated mice with the genetic disorder died much earlier, the researchers found.
Most importantly, the researchers say, the chemically-oriented process resulted in no off-target effects from treatment, which has been a major concern for other gene editing tools like CRISPR/Cas9, which can erroneously damage untargeted DNA and cause complications. Such collateral damage has limited CRISPR/Cas9’s therapeutic applications, they say.
The fetal research study was patterned after an earlier successful trial of the gene editing process in anemic adult mice. Bahal served as the lead author of the adult mice study, which was reported in Nature Communications in 2016. For both studies, Bahal designed and synthesized the next generation gamma PNAs used in the treatment. These modified PNAs exhibit enhanced binding to DNA, which provides better capacity for gene editing.
“These are not trivial molecules,” says Bahal. “They need very rigorous synthesis as well as strong quality control analysis.”
Working with diseased fetal tissue presented an enormous challenge for the researchers. The latest effort was a true collaborative process, Bahal says. The PNA molecules were synthesized in Glazer’s lab and the nanoparticles were formulated in the lab of W. Mark Saltzman, Yale University’s Goizueta Foundation Professor of Biomedical Engineering, Chemical & Environmental Engineering, & Physiology.
Professor Danith Ly of Carnegie Mellon University provided material for the gamma PNA molecules, and the team relied on the expertise of Yale pediatric surgeon Dr. David H. Stitelman in making the necessary fetal injections.
“Formulating the nanoparticles and injecting them in fetuses is a very sensitive technique,” says Bahal. “You have to be very careful that everything is pure and sterile, and the particles need to be delivered in an appropriate amount at just the right place so as not to cause toxicity. In utero injections with other gene editing tools are challenging, due to off-target damage.”
Treating the disorder at the fetal stage allowed the researchers to target important stem cells, which propagate the corrected DNA once they are treated, rather than DNA carrying the mutation.
“People who have thalassemia, they get sicker and sicker as they go on because they don’t have normal red blood cell function, and it gets harder to treat,” says Saltzman. “Here, we’re correcting the gene very early in development, so you see more benefits because they don’t get sick.”
Moving forward, the researchers plan to test the approach on other genetic disorders such as sickle cell anemia, cystic fibrosis, and Hurler disease.
Bahal says a recent grant he received from Cooley’s Anemia Foundation will be critical in allowing him to build resources in his new UConn lab to take the gene editing technology to the next level.
Beta thalassemia is caused by more than 200 different genetic point mutations. The successful results reported in the recent study only addressed those related to a beta globin gene mutation in individuals of Asian heritage, which was selected for the study, says Bahal.
“With this Cooley’s Anemia grant,” he says, “we will further advance this technology to fix other pivotal point mutations for beta thalassemia gene therapy.”
The research was funded by the Brain Research Foundation Scientific Innovations Award, the NIGMS Medical Scientist Training Program (GM07205), the National Heart, Lung, and Blood Institute (HL134252), the Ohse Research Grant, Yale School of Medicine, the American Pediatric Surgical Association Foundation Grant, and the DSF Charitable Foundation.


