Imagine having to drink a solution of cornstarch and water every few hours to survive, and that missing a dose even by 15 minutes could lead to seizures and death. This is how those with Glycogen Storage Disease Type Ia (GSD-Ia) live every day of their lives.
GSD is a metabolic disorder caused by an enzyme deficiency where the liver fails to break down glycogen into glucose, causing the body’s blood sugar levels to drop. The disorder is devastating, causing potential damage to kidneys and liver along with other serious side effects, including death.
GSD was almost always fatal until 1971, when it was discovered that continuous glucose therapy could prevent hypoglycemia. In 1982, cornstarch therapy was introduced as a slow-release form of glucose.
In 2018, after working toward a breakthrough for 20 years, Dr. David Weinstein, the then-director of the Glycogen Storage Disease Program at Connecticut Children’s and UConn Health, and his team at UConn Health administered the world’s first investigational gene therapy to a participant with Glycogen Storage Disease Type Ia (GSD-Ia) as part of the Phase 1/2 clinical trial.
When Weinstein departed UConn Health to research other rare diseases, Dr. Rebecca Riba-Wolman, director of the Glycogen Storage Disease Program at Connecticut Children’s and UConn Health and member of the Division of Pediatric Endocrinology at Connecticut Children’s, did not miss a beat in taking over the program and research her colleague and mentor started.
“The participants of the Phase 1/2 trial overall have seen a 70% decrease in cornstarch needs,” Riba-Wolman says. “Some participants who could previously not go more than four hours without cornstarch are now able to last from 12-15 hours, significantly changing their lives.
“Dr. Weinstein left me with an incredible team of people and I couldn’t do this without them,” she says. “Including co-principal investigator Dr. Karen Loechner, and nurses with GSD and ICU experience, the team also includes a metabolic dietician, Malaya Mount, who monitors the participant’s diet and cornstarch intake based on the data from their glucose monitor.”
Julie Vigil, administrative director, Department of Pediatrics at UConn Health has the passion and vision to make sure the inpatient clinical trial unit has the resources and ability to move this into an actual therapeutic option for children and young adults with GSD.
Leading up to the trial, numerous steps must take place, and Shaylee King, study coordinator, UConn Health has been instrumental in handling all the regulatory submissions and approvals as well as recruitment and care of subjects. King is joined by another study coordinator, Julieta Bonvin-Sallago, who, prior to coming to the United States, was a metabolically trained physician.
The Trial – Phase 3
You could feel the buzz of excitement in the air on Tuesday, Jan. 18 in the inpatient clinical trial unit at UConn John Dempsey Hospital, as the team that has been preparing for months were ready for the groundbreaking clinical trial participant to be the first in the world to receive the infusion of the potential gene therapy to kick off Phase 3 of the trial.
“This is very exciting. It’s a culmination of over 20 years of research initiated by our colleague Dr. David Weinstein, who was the visionary and who had the lasting capacity to move this from an idea to the animal model to trial that we are now in, Phase 3, and closer to bringing this innovative, life-changing gene therapy to the broader GSD community,” says Dr. Juan C. Salazar, Physician-in-Chief, Connecticut Children’s and Department Chair of Pediatrics at UConn School of Medicine. “To me, this is very exciting that together, UConn Health, the Department of Pediatrics at the UConn School of Medicine, in partnership with Connecticut Children’s, are able to be the first in the world to move to the new phase of the trial.
“The other thing that is remarkable is that we are doing this in the midst of COVID,” says Salazar. “UConn Health has the highest number of COVID cases that we have had at any given time, yet the leadership has made it possible to set up this unit here and continue this research.”
The ground-breaking clinical trial is being done in conjunction with the biopharmaceutical company Ultragenyx Pharmaceuticals, based in Novato, California. The trial’s primary goal is testing the safety and dosage of the investigational gene therapy for those with GSD-1a. This research has been generously supported by the local GSD community.
In the Phase 3 blinded control trial, 50% of the participants will receive the investigational product and 50% will have a placebo. No one involved in the trial will have access to the information identifying who has the product and who has the placebo.
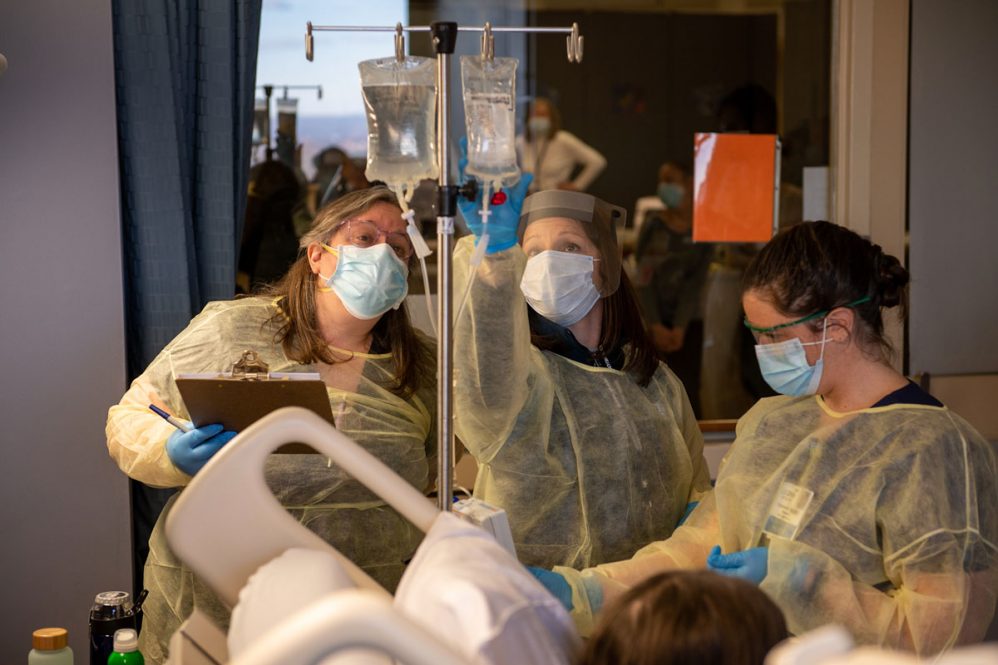
With parents by their side and family on FaceTime, as well as Riba-Wolman and nurses in the room, the participant – whose name and identity are being kept confidential to preserve the integrity of the double-blind trial – received an IV of the investigational product potentially that took about 45 minutes to administer. There were tears of joy as the work of so many came to fruition, and the prospect of changing the lives of the participant and other GSD patients hung in the air.
The gene therapy is carried through a participant’s bloodstream, by a naturally occurring virus, to the liver to replace deficient sugar enzymes caused by the disease and to jumpstart the body’s glucose control.
The participant will spend three days in the inpatient clinical trial unit to be monitored.
“Participating in the trial is a big undertaking and I admire the dedication it takes not just doing it for themselves, but that they are putting in the work to help other people as well,” says Riba-Wolman.
The participant will be gathering and reporting data daily including documenting what they are eating and how much cornstarch is being taken, as well as daily finger pricks to check the blood sugar. The Dexcom continuous glucose monitor will provide this information directly to the study coordinators.
Every three months the participant will come back to UConn Health for a fasting study.
The study will take two years, but after the first year, anyone receiving the placebo will be switched over to the investigational product.
Upon completion of Phase 3 of the trial, the findings will be published and peer-reviewed, with the ultimate goal of a treatment that will be widely available for all those who have GSD-Ia.
The Participant
Participating in a clinical research study is a brave and selfless act, and the participant in this trial embodies just that, as they have faced GSD for their entire life, with a positive attitude and grace beyond their years.
“I am always in awe of those willing to do these types of studies, including this participant getting the therapy,” says Salazar. “We are so grateful to the families and participants willing to step in and believe in us for a blinded trial where they don’t know if they are getting the placebo or the investigational product.”
The participant was first diagnosed at 5 months old. Their mother describes the participant as a generous soul and wise beyond their years.
“It was life-changing when we met Dr. David Weinstein in 2001 when [the participant] was 7-8 months old,” says the participant’s father.
The participant had a feeding tube when they were a baby to make sure they had a constant source of glucose. As common with GSD babies, the lack of use of their mouth then required intense speech therapy to teach them how to eat.
Every four hours, they have the cornstarch solution and dosing has changed throughout the years.
“To think that a common household item almost everyone has in their cabinet like uncooked cornstarch is their medicine and taken at the correct volume, along a strict regimen is lifesaving, literally lifesaving,” says the father.
Growing up as a child, the parents would have to set alarms to wake up throughout the night to give their child cornstarch. If they overslept, the child’s blood sugar would drop, and the participant would become hypoglycemic, having shakiness and severe reaction.
Now as a young adult, the participant takes Glycosade, approved by the FDA in 2012 for the dietary management of GSD. Glycosade is a “superstarch” that can maintain blood sugar levels for 7-8 hours overnight, allowing GSD patients and their families to get a full night of sleep.
The participant has also used a Dexcom continuous glucose monitor, which connects to their cell phone, as well as to the cell phone of roommates and family members, so that if the blood sugar level drops others are aware as well.
“That has been a real lifesaver,” says the participant. As part of the study, this option has been replaced with a blood sugar monitor that study investigators will monitor.
“Participating in the study is a dedication not just for themselves; they are putting in the work for other people,” says Riba-Wolman.
GSD not only affects the child who has the disease, but also deeply affects parents and family as well. The parents agree that being completely open about the participant’s GSD has helped them and others understand.
“It’s a team effort. We have the support of family and the close GSD community,” says the participant’s mother.
“Over many years, it’s been a big weight for [the participant] and our family to carry, and this gene therapy, even though it isn’t a cure, it alleviates the amount of cornstarch they have to take and makes the symptoms less severe. That’s a huge plus in [the participant’s] life and our lives too,” says the participant’s mother.
“I think gene therapy will be amazing. Even a tiny reduction in the overall cornstarch I consume will be life-changing, including less mental stress, less physical stress, and an overall better life,” says the participant.
The Clinical Trials Unit through the UConn Health Department of Pediatrics offers research for all ages, not just children and adolescents. We specialize in gene therapy and infusion trials, we currently have multiple rare disease trials. UConn Health has partnered with Connecticut Children’s to be able to provide the best research and safest facility for our subjects.
The highly-skilled, board-certified pediatric endocrinologists at Connecticut Children’s provide world-class integrated health care to infants, children and teens with a broad range of endocrine diseases, metabolic abnormalities and hormone conditions.
If you would like to learn more and see if you or a loved one may qualify for one of our clinical trials please email us at PediatricClinicalTrials@uchc.edu.