
A crowd of families presses into Stormy Chamberlain’s lab. The adults have hair of every color, but most of the kids are blond. Everyone waits their turn to look through a microscope, where clumps of cells float into view like round, yellowish blobs.
What are we looking at? someone asks.
“Brain bits,” Chamberlain says.
Chamberlain is a developmental geneticist at UConn Health who researches the genetic basis of brain disorders. The families in her lab today have a personal stake in her work, because each of them has a child with Angelman Syndrome.
People with Angelman Syndrome have delayed development, usually cannot speak more than a few words, and tend to be unusually fair with blond hair and blue eyes. They also tend to be happy, and smile a lot.
“She can’t talk, but she’s like a social butterfly,” Jocelyne Braffith says about her four-year-old. Braffith’s daughter has what geneticists call a deletion. A single gene, UBE3A, is deleted on the 15th chromosome she inherited from her mother. She has a normal copy from her father, but for reasons no one yet understands, only the mother’s copy is used in the brain. Because her mother’s copy of UBE3A is missing in action, she has Angelman Syndrome.
A tall blond girl named Katie wanders past and shows us a test tube Chamberlain gave her.
“Wow, that’s cool,” Braffith says. Katie grins and taps the test tube knowingly, then walks off to show someone else. Katie has a different form of Angelman Syndrome than Braffith’s daughter. Instead of a deletion, she has an imprinting error. Her mother’s copy of UBE3A is right where it should be on the 15th chromosome, but it’s imprinted to behave like her father’s. So it doesn’t turn on in Katie’s brain, and she doesn’t talk.
Chamberlain and her students want to know why. Why is only the mother’s copy used in certain parts of the brain? Can the father’s copy be turned on instead? How early on does Angelman brain development start going wrong?

UConn researcher Stormy Chamberlain studies the genetic basis of brain disorders, but she never forgets the families who have a personal stake in her work.
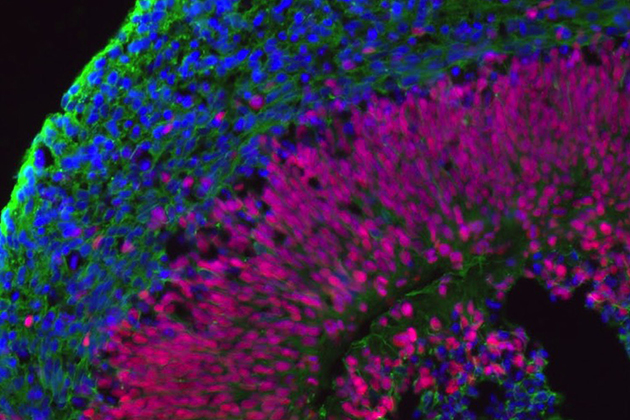
Which leads us to the brain bits. Angelman families donated blood or skin cells to Chamberlain’s lab. The researcher and her students turned those cells into stem cells and then encouraged those cells to grow into brain tissue – neural epithelium. The neural epithelial cells arrange themselves into little balls, like tiny brains. The researchers study the brain bits in hopes of pinpointing exactly when an Angelman brain stops developing normally.
Everyone in the group has a good look at the brain bits. Some people even try to photograph them, angling their phones carefully through the microscope’s eyepiece, and one man jokes that he wants one embedded in plastic as a souvenir. Then the families file out of the lab and gather for a question-and-answer session.
Carissa Sirois, a second-year Ph.D. student, describes how she’s using the Angelman cells to do a side-by-side comparison with normal brain cells. She’s going to grow out a line of Angelman cells, and then a second line of cells identical to the first except that she’ll have corrected the mutation in the UBE3A gene so they no longer have Angelman Syndrome. Then she’ll carefully track the RNAs produced by both cell lines. RNAs are ribonucleic acids, the precursors to proteins and other stuff made by cells. Any RNA produced by the normal cells that is missing from Angelman cells could be a clue toward a potential treatment.
But treatment is a tricky thing to talk about when it comes to Angelman Syndrome. It is not known whether the brain differences in Angelman Syndrome can be helped or cured once a person is old enough to display symptoms. A team in the Netherlands has used gene editing to add UBE3A back to Angelman mice, but it only helps if it is given to the mice before they are three weeks old.
And Angelman Syndrome is rare enough that it isn’t routinely tested for during prenatal or newborn care. Most families don’t get a diagnosis until their child is at least a year old, and unless a test is done, doctors rarely recognize it. They often give a generic diagnosis of autism instead.
It’s helpful to have a specific diagnosis, says Barbara Dell, whose son Gabriel is affected. “There’s a framework [to Angelman]. It gives you a clearer picture of what to expect. You know my kid is never going to go to college. But it’s not degenerative, so we know what he gains, he’ll keep. We know he won’t be able to live independently. So we can plan.”
Dell and the other families are raising money for research through the Angelman Walk in Manchester on May 16. The money will go to the Angelman Syndrome Foundation, a national organization that funds research into the disorder.
Chamberlain attends the walk every year, and sometimes other UConn researchers go with her.
As the question-and-answer session winds down, Chamberlain tells a story about the Angelman Walk a few years ago, when she first met Katie. She picked a flower and gave it to her. Katie sniffed it rapturously and then tossed it away. Chamberlain gave her another one, and then another one, and she kept sniffing them and then tossing them. It became a running joke as they walked.
“Katie has a sense of humor,” Chamberlain says. “You meet people with Angelman, there’s a whole person in there, and you really want to help the families.”
It’s why she likes the families coming to the lab, and gets her graduate students to come out and meet them.”Knowing that real people have a stake in the research,” she adds, “is one of the things that keeps us going.”


