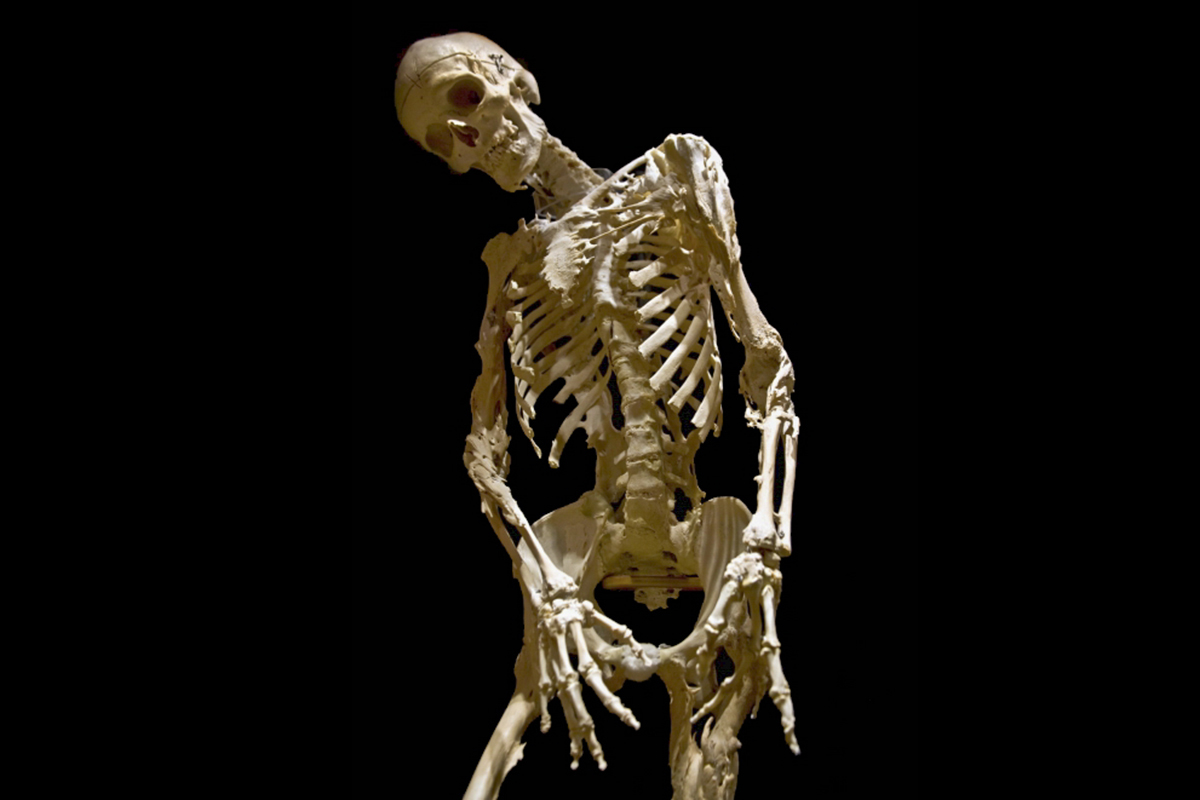
By the time he died in 1973, Harry Eastlack had two skeletons; the one he was born with, and the one that grew around it, gradually encasing him in a prison of his own bone. Forty-five years later, UConn researchers have shown in the Feb. 2, 2018 of Nature Communications that a certain type of cell is responsible for Eastlack’s disease. Their results could help others with this disease, as well as hint at why millions of people suffer from bony growths after sports and deep tissue injuries.
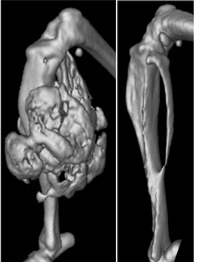
Fibrodysplasia ossificans progressiva (FOP) affects only 1 person in 2 million. It’s caused by a spontaneous mutation that affects only a single protein. But the effect is devastating.
“In FOP, the most mild injuries – a childhood vaccination, a bruise – triggers massive bone growth. And nothing can be done about it. It can’t be surgically removed, because that triggers more bone,” says UConn stem cell biologist David Goldhamer. “If bone grows across a joint, that joint locks, and it will never move again.” Inevitably, FOP sufferers grow more and more bone, slowly getting locked in this secondary skeleton. Many die in midlife, often of respiratory problems due to their inability to breathe well. Eastlack died at age 39 from pneumonia.
Goldhamer began studying FOP when he was at the University of Pennsylvania and a doctor who treated FOP patients approached him. What is special about muscle tissue, the doctor asked. Why does muscle give rise to these bony growths? After meeting FOP patients and their families, Goldhamer was inspired to use his expertise in muscle biology to address these questions. He has researched FOP ever since, and he finally has answers to the doctor’s questions.
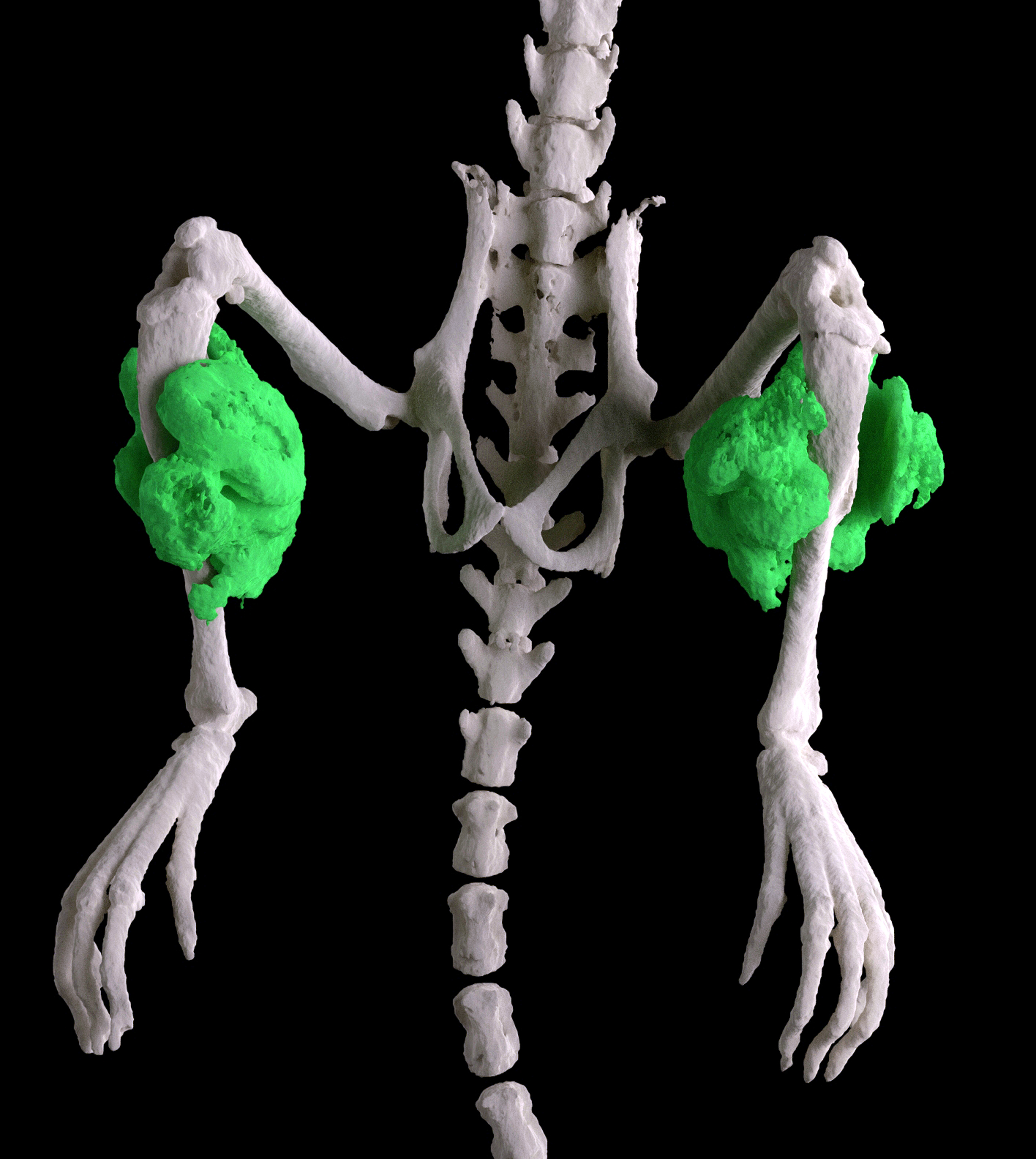
The mystery bone-making culprit, it turns out, isn’t related to muscle. Instead, it’s a type of cell called Fibro/Adipogenic Progenitors (FAPs). FAPs hang out in muscle tissue, but it’s not entirely clear what they do. No one had any idea they were capable of making cartilage and bone.
Goldhamer, post doc John Lees-Shepard, and assistant research professor Masakazu Yamamoto at UConn, as well as colleagues at the University of Michigan and Alexion Pharmaceuticals, examined FAPs with a mutation in the ACVR1 protein. University of Pennsylvania scientists had already identified the mutation as the cause of FOP. What Goldhamer and his colleague showed is what it does: it alters a single type of protein that acts as a receptor on the surface of FAP cells, and this mutant receptor somehow tells FAPs to develop into cartilage and bone at injury sites.
Now Goldhamer and his colleagues are trying to understand the basic biology of how and why the disease progresses, in the hopes that future FOP patients won’t meet the same fate as poor Harry Eastlack.


